双环[1.1.0]丁烷(BCBs)因其C1-C3键的高反应性成为有机合成中的多功能构建砌块。然而,将BCBs应用于螺环骨架的合成仍然面临挑战。螺环丁烷因其多样的生物活性,在药物化学领域引起了广泛关注。因此,通过简单高效的方法合成6,7-二氮杂螺[3.4]辛烷具有重要的科学意义。陕西师范大学薛东/董建洋团队基于前期研究的工作基础(Chem. Sci. 2025,16, 4654; Sci. China. Chem. 2024, 67, 3389; Green. Chem. 2024, 26, 5531; Org. Lett. 2025, 27, 3571; Org. Lett. 2024, 26, 6230; Org. Lett. 2024, 26, 7026; Org. Lett. 2024, 26, 9276; Org. Lett. 2024, 26, 9311;),报道了钪催化BCBs与偶氮甲碱亚胺螺环化合成6,7-二氮杂螺[3.4]辛烷(图1),相关研究成果发表于Chem. Sci. 2025, 16, 12189.
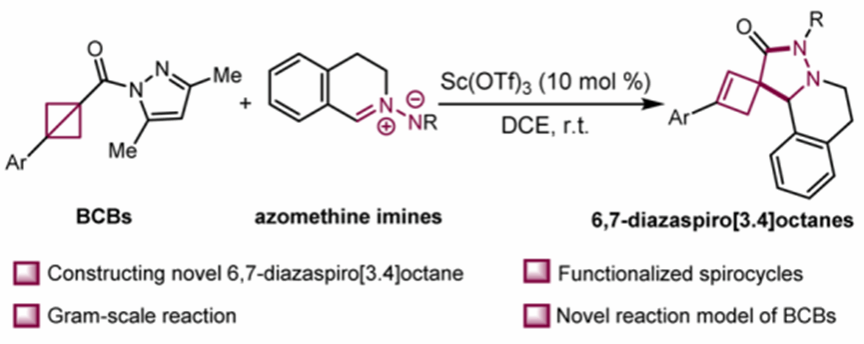
图1. 钪催化BCBs与偶氮甲碱亚胺的螺环化反应
BCBs是由两个环丙烷组成的张力环,在有机合成、药物化学和化学生物学中有着广泛的应用。这主要是由于一方面BCBs表现出高达67 kcal/mol的张力能,具有很高的反应活性;另一方面是BCBs桥头间C1-C3键的独特反应性,BCBs中心C1-C3键具有96%的p轨道特征,赋予其类似π键的反应活性。基于BCBs特殊的反应活性,发展了通过张力释放开环策略合成含四元环(环丁烷和环丁烯)结构的方法。已使用包括亲核试剂、自由基、亲电试剂和过渡金属催化剂成功实现这种高能键的环张力释放,合成了系列官能团化的环丁烷和环丁烯(图2A)。除了开环反应外,BCBs通过(3+n)环加成反应成为构建双环[n.1.1]烷烃的通用策略(图2A)。此外,通过金属化BCBs的酸性C-H键,进一步拓宽了骨架多样化(图2A)。尽管BCBs展现出丰富的化学活性,开发螺环化策略将能快速获得功能化的螺环丁烷衍生物。在过去的几十年里,螺环骨架已成为药物化学领域的重要药效团,众多螺环化合物因其显著的生物活性而被发现。值得注意的是,含有螺[3.4]辛烷的化合物因其显著的抗癌活性而尤为突出(图2B)。尽管螺环化合物有很大的潜力,但螺环化合物的合成方法十分有限。因此,亟待开发更简单高效、普适性更强的策略来合成螺环骨架。
基于BCBs的高反应活性和在螺环骨架构建方面的空缺,作者拟通过释放BCBs的张力能驱动其合成螺[3.4]辛烷。C,N-环状偶氮甲碱亚胺类化合物作为1,3-偶极子在环加成反应中具有高反应性,但其研究主要集中于通过[3+2]、[3+3]和[3+4]环加成构建含氮杂环。目前,利用C,N-环状偶氮甲碱亚胺通过螺环化反应合成螺环含氮杂环尚未见报道。因此作者设想能否利用路易斯酸活化BCBs生成碳负离子中间体A。该中间体随后可与C,N-环状偶氮甲碱亚胺的亲电部分反应形成中间体B。最终,B通过分子内亲核取代生成螺环丁烷(图2C)。
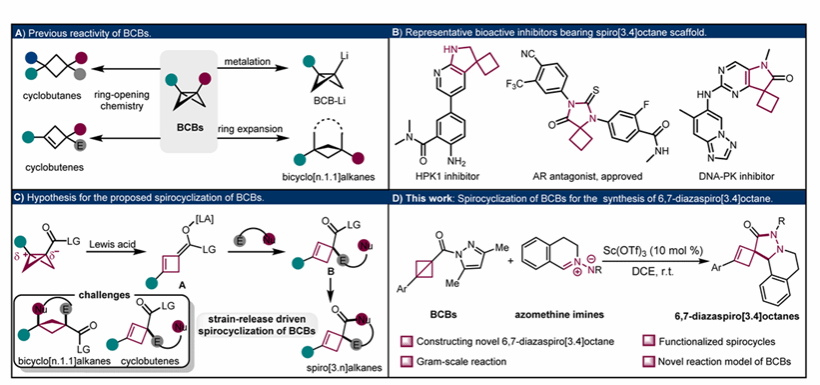
图2. BCB环加成反应合成桥环化合物
通过对反应条件进行筛选,作者发现在Sc(OTf)3催化下,使用DCE作为反应溶剂,在常温条件下反应能够以87%的收率,1.7:1的非对映选择性得到螺[3.4]辛烷产物。随后,作者首先对双环[1.1.0]丁烷(BCB)底物范围进行了考察(见图3),该反应对于各种各样取代基的双环[1.1.0]丁烷(BCB)具有良好的适用性,均能以中等至良好的收率得到目标产物。紧接着,作者考察了偶氮甲碱亚胺的底物适用范围(见图3)。该反应具有良好的适用性,均能以中等至良好的收率得到目标产物。在成功建立了BCBs与偶氮甲基亚胺的反应后,作者希望能够实现该反应的不对称螺环化反应。当使用BINOL衍生的CPA作为催化剂,1,4-二氧杂环己烷作为溶剂时(见图3),能以66%的产率,1.7:1的d.r.值,82%/63% ee得到目标产物3a。
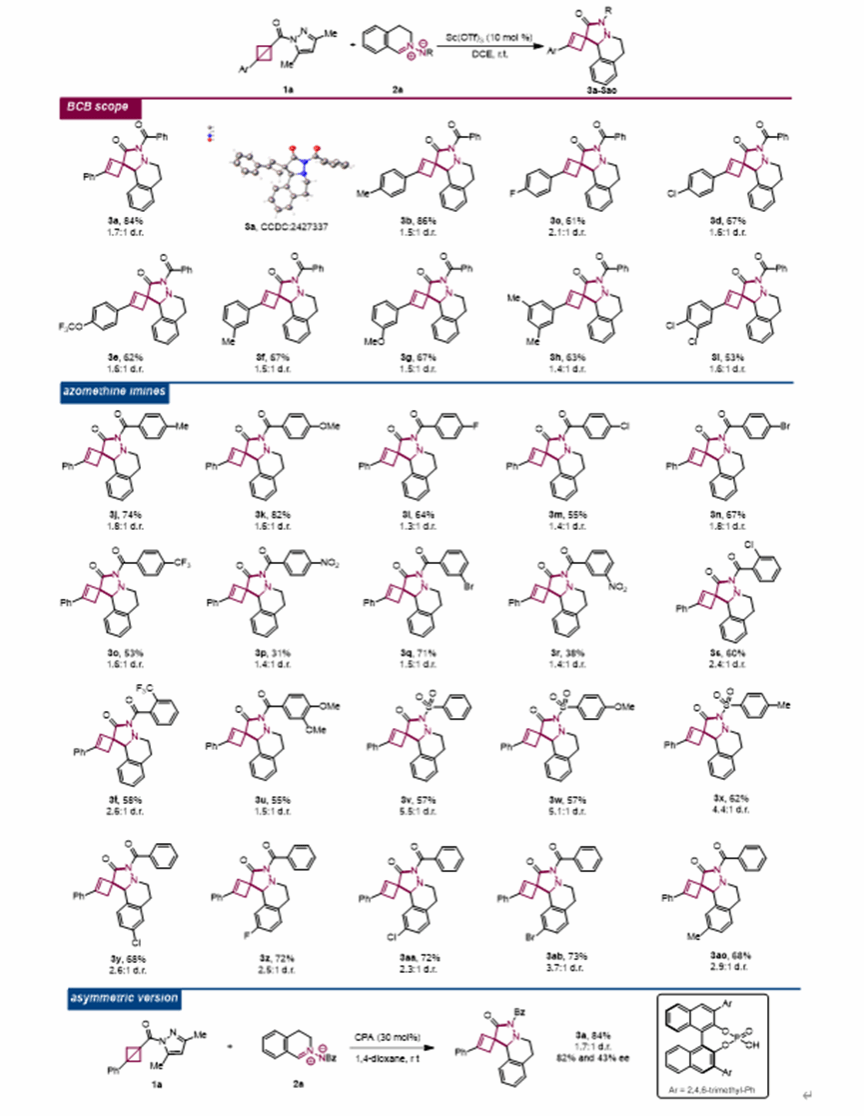
图3. 底物拓展
同时,为了验证该反应的实用性,作者进行了克级规模的放大反应,以4.9:1的d.r.值,50%的产率得到了目标产物3v。此外,作者对3v进行了转化(见图4)。具体而言,使用Mg/MeOH对3v进行脱保护,以4.9:1的d.r.值,69%的产率得到了化合物4。此外,3v中的羰基被还原成相应的醇,以2.4:1的d.r.值54%的产率生成了化合物5。随后,3v在还原条件下使用Pd/C作为催化剂成功进行了氢化反应,以1:1的d.r.值64%的产率得到了产物6。最后,通过3v与NBS在水的存在下反应,合成了卤代醇7。
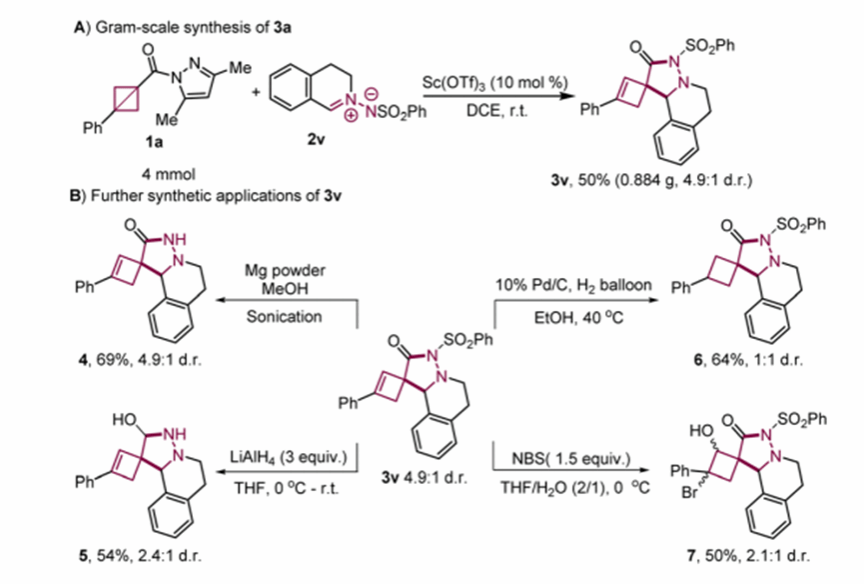
图4. 克级反应和衍生转化
最后,作者进一步研究了反应机制。首先,在没有BCB(1a)的情况下,2a在标准条件下未形成任何产物,大量起始材料仍存在(图5A)。然而,当1a在相同条件下,且没有(2a)时,以10%的产率获得了环丁烯8(图5B),这表明1a可能直接被Sc(OTf)3活化,形成阳离子中间体。最后,向含有1a和2a的反应混合物中加入自由基捕获剂TEMPO,并未显著影响3a的产率(图5C),这表明该反应不经历自由基途径。基于这些机理实验,作者提出了图6所示的机制。首先,底物1a通过与Sc(OTf)3配位至羰基,形成阳离子中间体I。中间体I的消除生成中间体II。随后,中间体II通过中间体III与底物2a发生亲核加成反应,形成中间体IV。这一关键中间体经历取代反应,氮原子攻击吡唑酰胺基团,生成所需的6,7-二氮杂螺[3.4]辛烷产物3a,并使Sc(OTf)3催化剂再生。
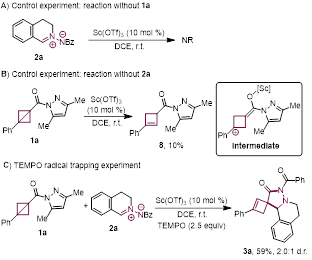
图5. 机理验证实验
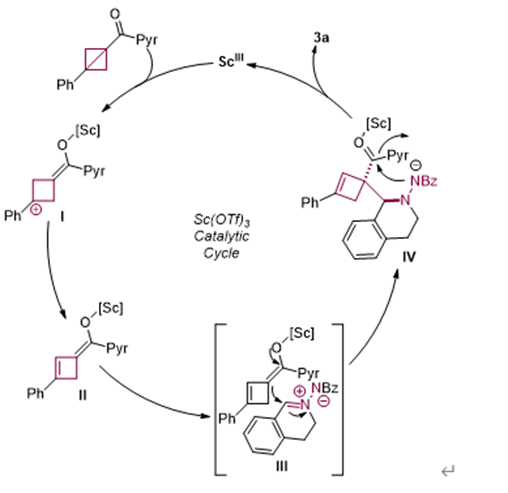
图6. 反应机理
作者报道了一种Sc(OTf)3催化的BCBs与C,N-环状偶氮甲碱亚胺的螺环化反应策略,成功构建了具有重要合成价值的6,7-二氮杂螺[3.4]辛烷骨架。通过克级规模反应和产物的后续转化,证明了这种方法的合成实用性。值得注意的是,这种方法不仅为合成具有潜在药理活性的新型6,7-二氮杂螺[3.4]辛烷衍生物提供了关键技术支撑,更拓展了BCBs类化合物螺环化反应的方法学工具库,为螺环化合物的合成提供新思路。
上述成果以Strain-release driven spirocyclization of bicyclo [1.1.0]butanes: access to 6,7-diazaspiro[3.4] octanes为题发表在Chem. Sci., 2025, 16, 12189。
陕西师范大学薛东教授、董建洋副研究员为共同通讯作者,第一作者为博士研究生江琴。上述研究工作得到了国家自然科学基金、中央高校基本科研业务费的资助。同时,感谢实验中心樊娟、孙华明和郭新爱老师和袁书培老师在实验数据和机理研究方面提供的实验技术支持。
供稿人:董建洋
 师大主页
师大主页
 手机版
手机版
 English
English